2M.1 FULLSTACKS - Implementation of TOPMed RNA-Seq pipeline within Globus Genomics using FAIR principles
Table of Contents
- Introduction
- Topmed RNA-Seq workflow
- Integration of Minids and BDBags
- End-to-end analysis using BDBags as inputs
- List of BDBags
Introduction
This README describes the implementation of TOPMed RNA-Seq analysis pipeline that uses BDBags and MINID within a Galaxy based Globus Genomics (GG) platform to support FAIR (Findable, Accessible, Interoperable, Reusable) research. We have implemented specific tools within GG that automate the use of MINIDs representing input databags and generate output BDBags along with provenance and performance metric that can be used to validate reproducibility.
Topmed RNA-Seq workflow
We selected the TOPMed RNA-Seq pipeline as described in detail at: https://github.com/broadinstitute/gtex-pipeline/blob/master/TOPMed_RNAseq_pipeline.md
Summary
For each input sample, this RNA-Seq pipeline generates:
- Aligned RNA-Seq reads (BAM format)
- Transcriptome BAM generated by STAR
- QC Metrics on the aligned reads
- Gene-level expression quantifications based on a collapsed version of a reference transcript annotation, provided as read counts and TPM.
- Transcript-level expression quantifications, provided as TPM, expected read counts, and isoform percentages
Components
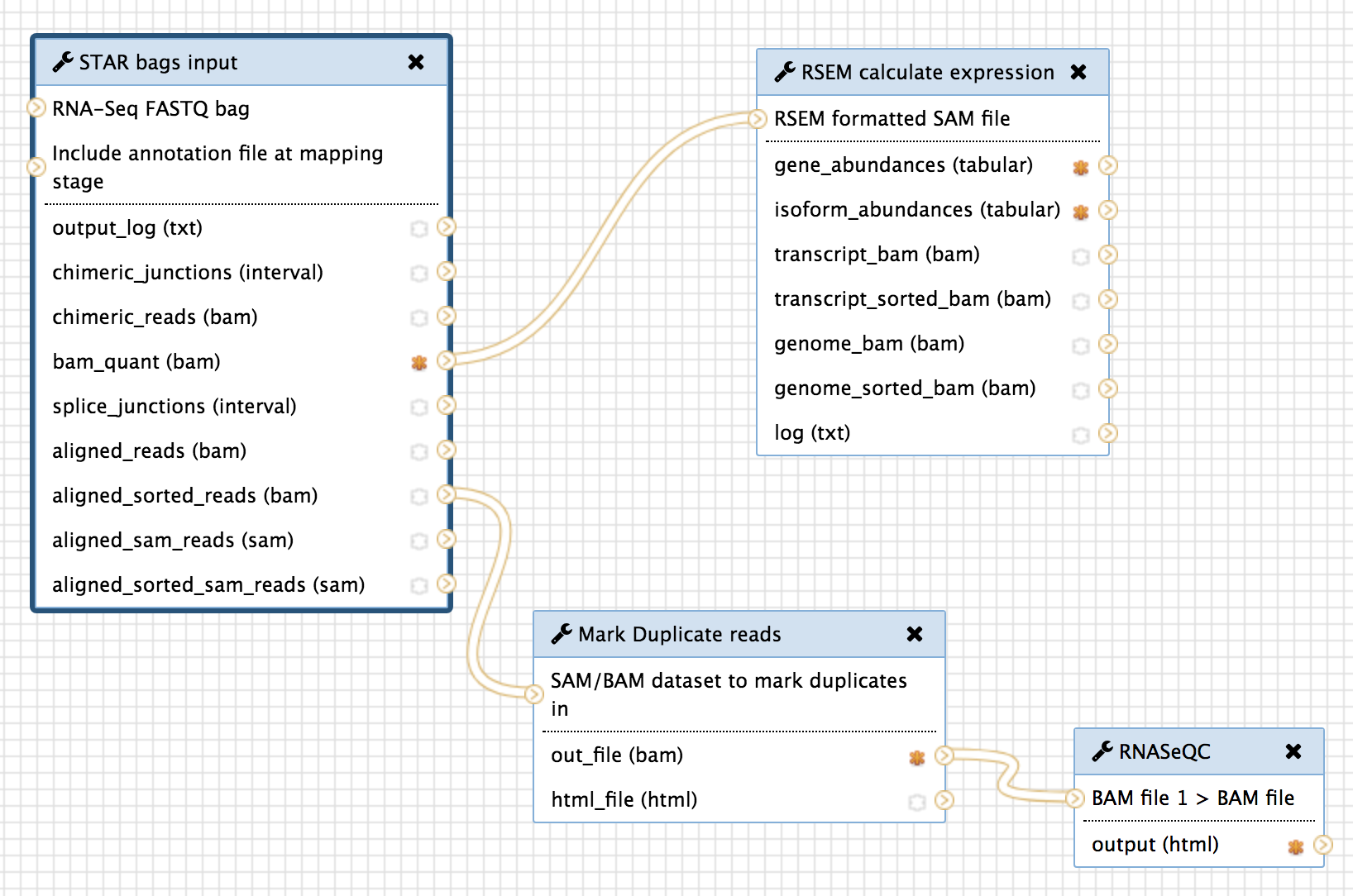
The figure below describes the RNA-Seq pipeline and its components:
We have wrapped the following versions of tools within Globus Genomics - Galaxy and we have made all the wrappers available within the Tools BDBag with MINID: ark:/57799/b9t690
The pipeline uses the following individual tools:
- Alignment: STAR 2.5.3a
- Post-processing: Picard 2.9.0 MarkDuplicates
- Gene quantification and quality control: RNA-SeQC 1.1.9
- Transcript quantification: RSEM 1.3.0
- Utilities: SAMtools 1.6 and HTSlib 1.6
Reference files
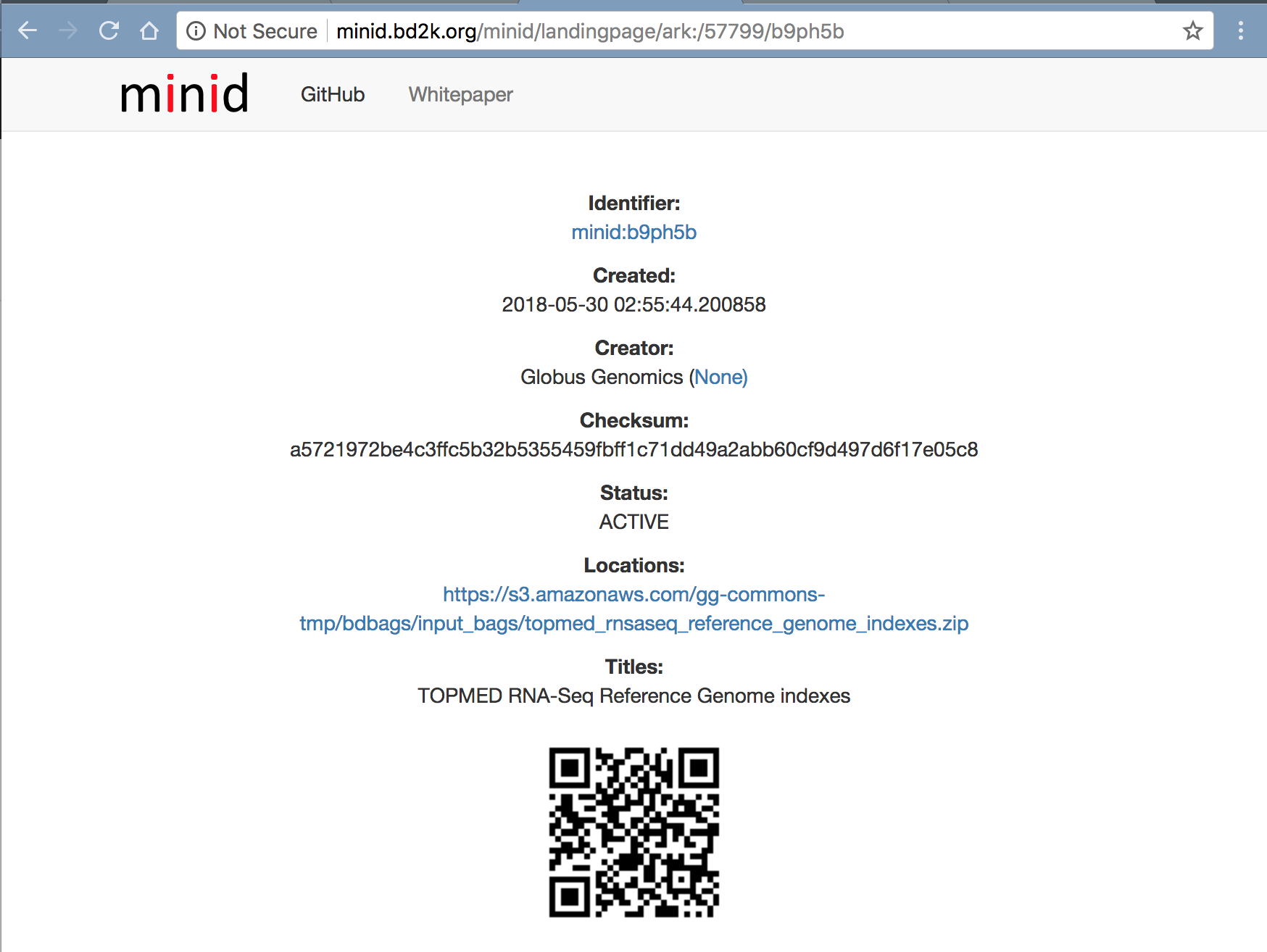
The tools listed above require Reference genomes. Following GRCh38 reference genome are used currently. We have created a References BDBag with all the reference databases in it. It can be accessed using the MINID: ark:/57799/b9ph5b.
- Reference genome for RNA-Seq alignment using STAR (contains .fasta, .fai, and .dict files): Homo_sapiens_assembly38_noALT_noHLA_noDecoy_ERCC.tar.gz
- Collapsed gene model GTF: gencode.v26.GRCh38.ERCC.genes.gtf.gz
- STAR index database: STAR_genome_GRCh38_noALT_noHLA_noDecoy_ERCC_v26_oh100.tar.gz
- RSEM reference database: rsem_reference_GRCh38_gencode26_ercc.tar.gz
Integration of MINIDs and BDBags
We have added new tools within Globus Genomics to support the use of MINIDs and BDBags. We have specifically added two tools - 1) Get BDBag using MINID, and 2) Create BDBag and MINID
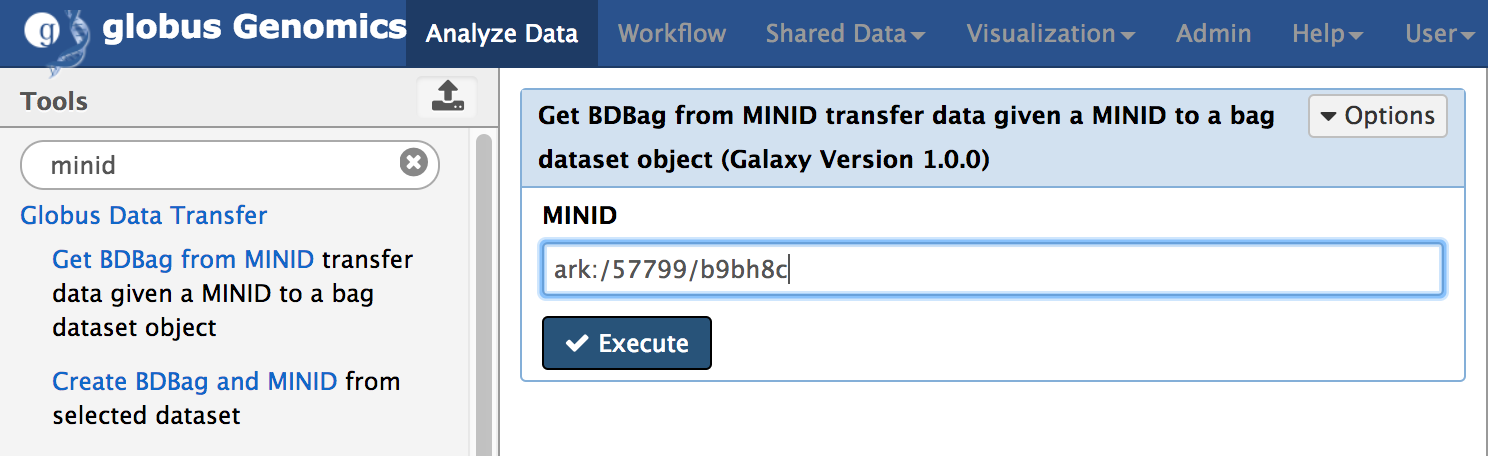
Get BDBag using MINID
Get BDBag using MINID is a new Galaxy tool we have added to GG that downloads all the data within the BDBag and adds it to Galaxy history. The following figure shows the tool within GG.
Publish results using BDBag and MINID
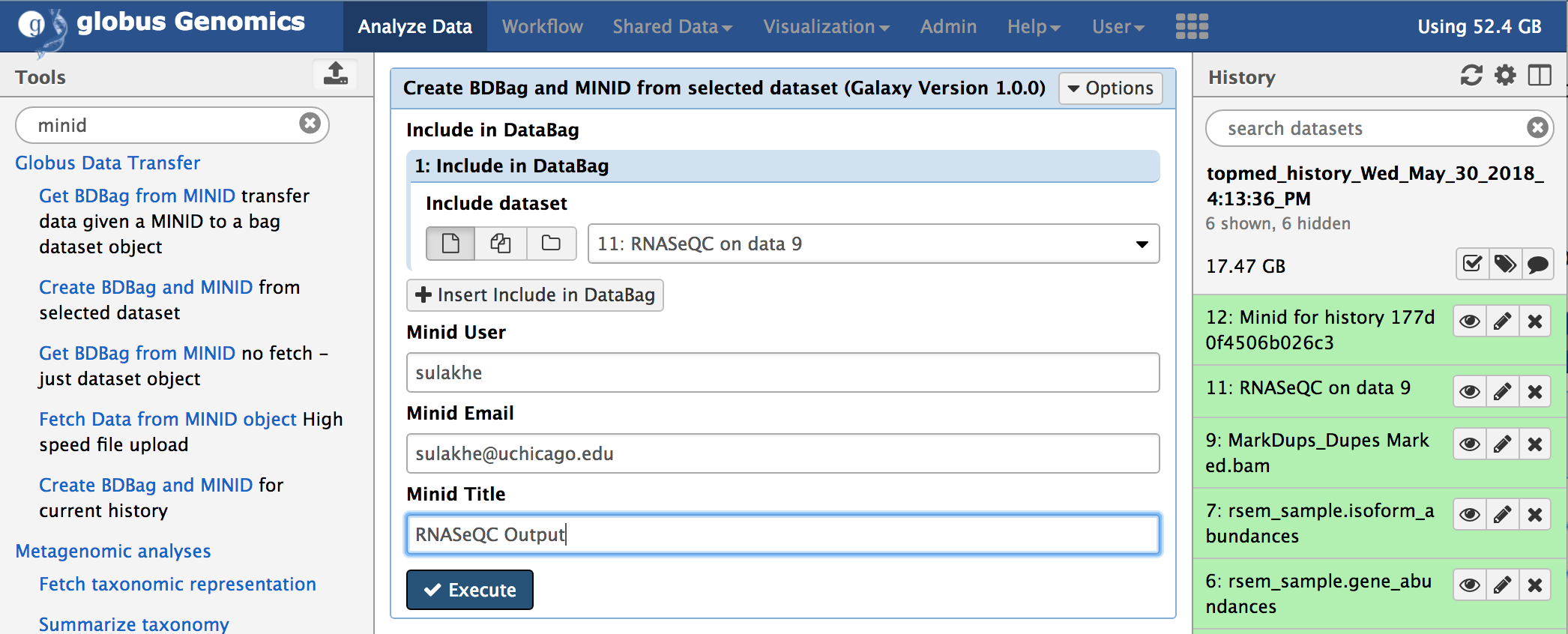
Create BDBag and MINID is a tool takes any history items or outputs of the tools and creates a BDBag and publishes it. It returns a MINID referencing the BDBag created. The following figure shows the tool within GG.
Integrate BDBags and Minids with Topmed RNA-Seq workflow
In order to provide an end-to-end automation of running the TOPMed RNA-Seq pipeline by using a MINID as input, we integrated the two tools within the RNA-Seq workflow shown above in Section 2/Components. The resulting workflow is shown below:
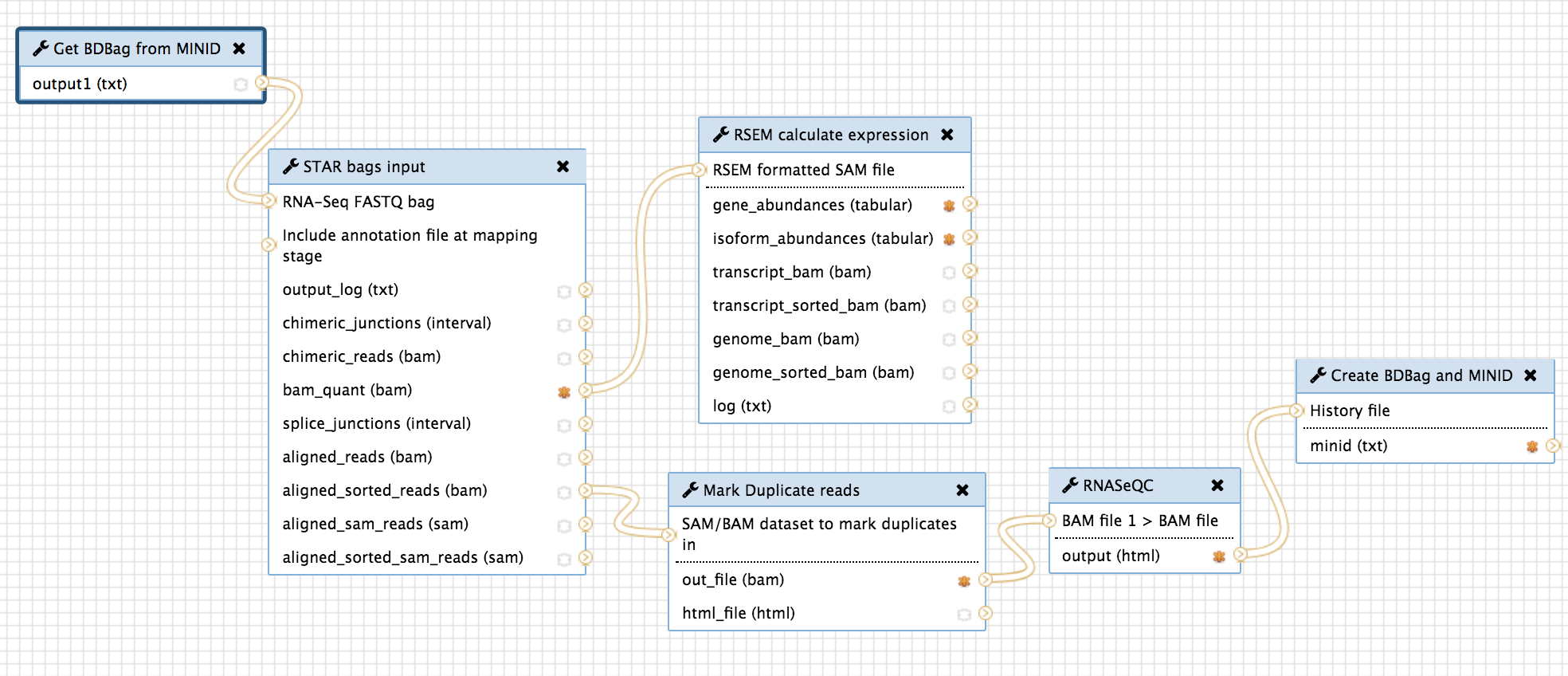
“Get BDBag using MINID” transfers all the data referenced by the MINID and feeds it into the first tool of the pipeline for analysis.
And the last step in the pipeline, “Create BDBag and MINID” takes the outputs of the workflow that are marked (orange stars in the above screenshot) and creates a new BDBag and generates a MINID for the bag. In addition to capturing the outputs of the analysis, this tools also collects provenance data in the form of actual command-lines with arguments that were used to run the tools, and performance metrics in the form of times taken to run the tools, and adds them to the new BDBag it creates. And example of the output BDBag is provided below under section 5(ii).
End-to-end analysis using BDBags as inputs
In this section we will describe how we can easily access the input data using a MINID and a pipeline required to run a RNA-Seq analysis and then validate if the results are as expected. Using Globus Genomics we will demonstrate how we can run this RNA-Seq pipeline using an input MINID in different modes. We will also describe how the outputs generated by the users can be validated and compared to ensure reproducibility of the results using BDBag infrastructure.
Authentication and Authorization
Authentication and Authorization to access the NIH-Commons Globus Genomics instance is a two step process.
- Create Globus ID: Create a Globus account at https://www.globusid.org/. The NIH-Commons Globus Genomics instance is made available to all the users with a Globus ID.
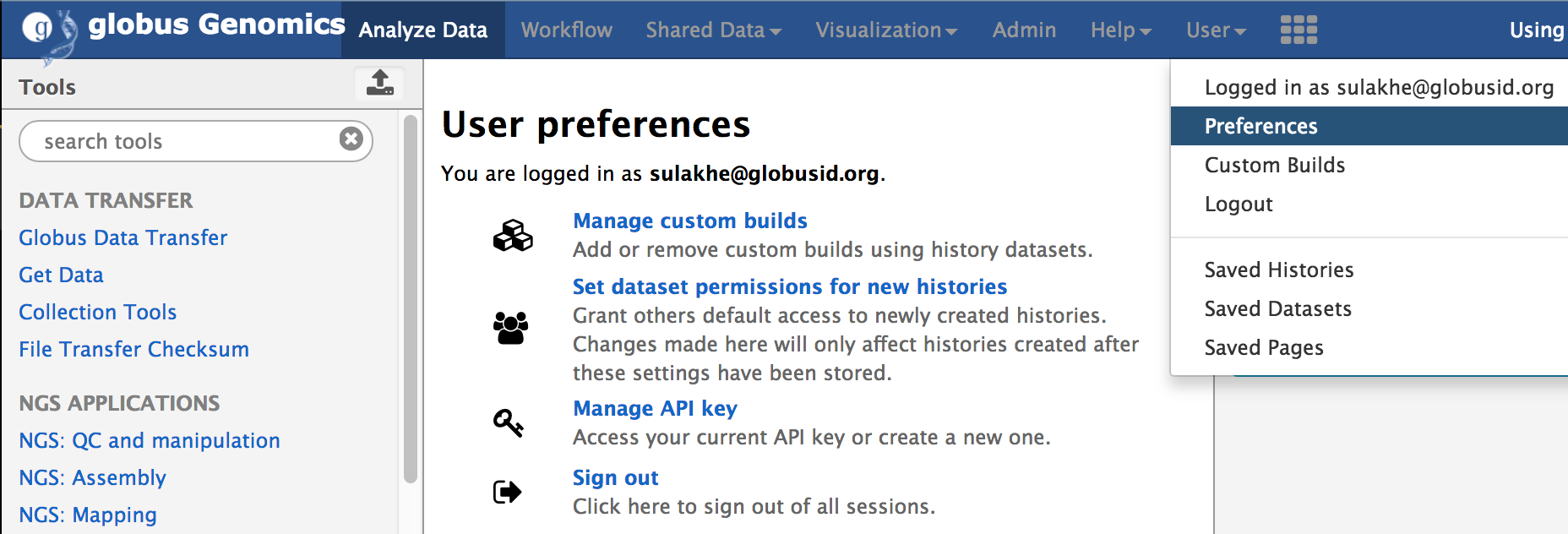
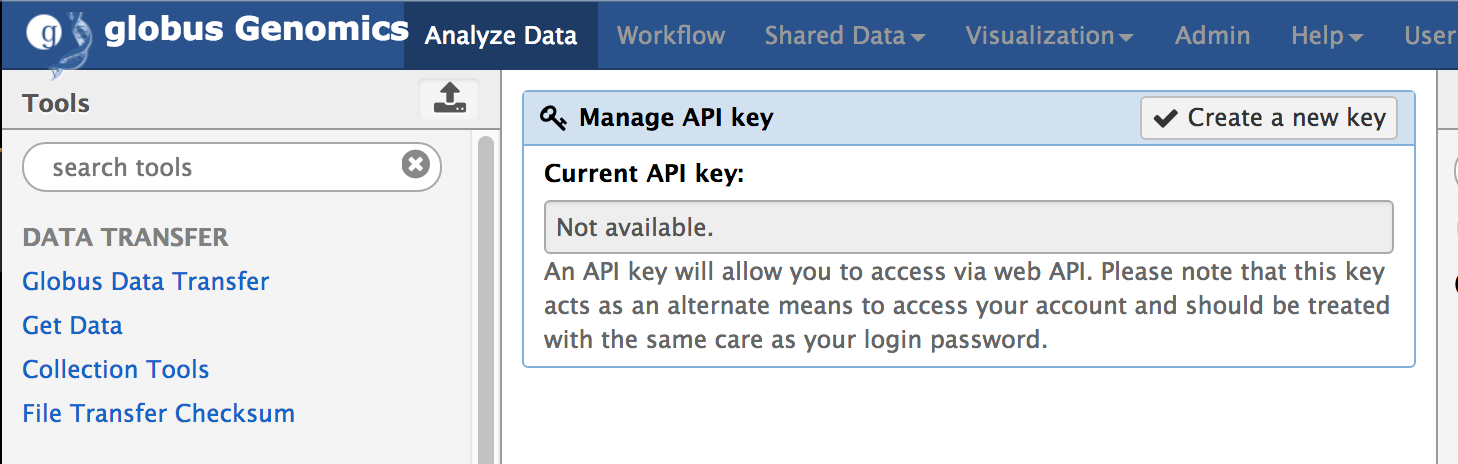
- Create Access API Key: Using the Globus ID, login to http://nihcommons.globusgenomics.org and from the top menu, go to User Preferences and click on “Manage API Key”, as shown below. If you don’t yet have a key, click on “Create new Key” button to create a new access key.
An API key will allow you to access via web API. Please note that this key acts as an alternate means to access your account and should be treated with the same care as your login password.
Using Python API
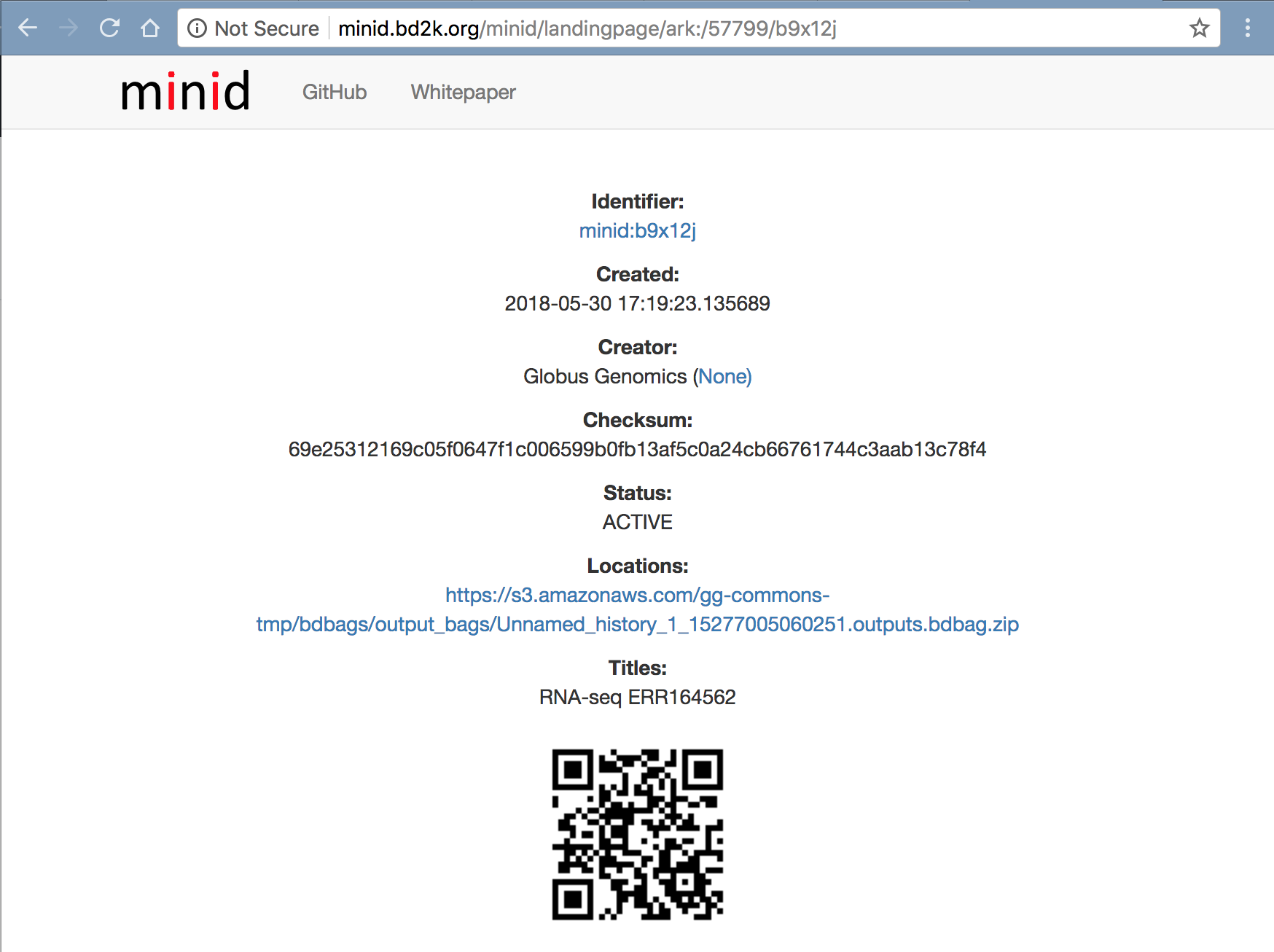
We have created a python script that uses Galaxy’s Bioblend API to run the workflow. It takes the MINID as input and run the workflow, and polls the status until the analysis is completed. After completion, the script returns a MINID for the outputs BDBag created at the end of the analysis. Each analysis creates a new outputs BDBag. Users can use the returned MINID to compare the outputs against the Outputs BDBag with MINID ark:/57799/b9x12j shown below to verify the reproducibility of the results.
Note: The python script requires the Access API Key generated in Step 2 above under Authentication and Authorization section.
We have created a MINID for the Python script at: http://minid.bd2k.org/minid/landingpage/ark:/57799/b9n97q
The Python script can run as follows:
- Create a Virtual Env:
$> virtualenv bioblend_env $> source bioblend_env/bin/activate - Install Bioblend Module
$> pip install bioblend - Download the Python script (using the Minid: http://minid.bd2k.org/minid/landingpage/ark:/57799/b92h64 )
```
$> wget https://raw.githubusercontent.com/fair-research/fair-genomics/master/submit_topmed_rna_seq.py
$> python submit_topmed_rna_seq.py –help
Usage: submit_topmed_rnaseq_workflow.py -k
--input-minid
Options: -h, –help show this help message and exit -k API_KEY, –key=API_KEY User API Key -m INPUT_MINID, –input-minid=INPUT_MINID Input MINID containing BDBag of paired-ended fastq files
$> python submit_topmed_rna_seq.py -k xxxxxxxxxxxxxxxxxx -m ark:/57799/b9bh8c SUBMITTED ark:/57799/b9bh8c imported: RNA-Seq-Gtex-stage1-v2.0-bags_transfer 801d769120b6adcb topmed_history_Fri_Jun_01_2018_12:13:21_AM 29015fce37680443 Workflow running: Fri_Jun_01_2018_12:13:23_AM ………
### Using Globus Genomics Web UI
**Step-1: Login:**
Using Globus ID, login to the NIH-Commons Globus Genomics instance at: https://nihcommons.globusgenomics.org
**Step-2: Create Access API Key**
If you have not already done it (as per Authentication and Authorization section). Please create the access API Key from "User->Preferences->Manage API Key" in the top menu.
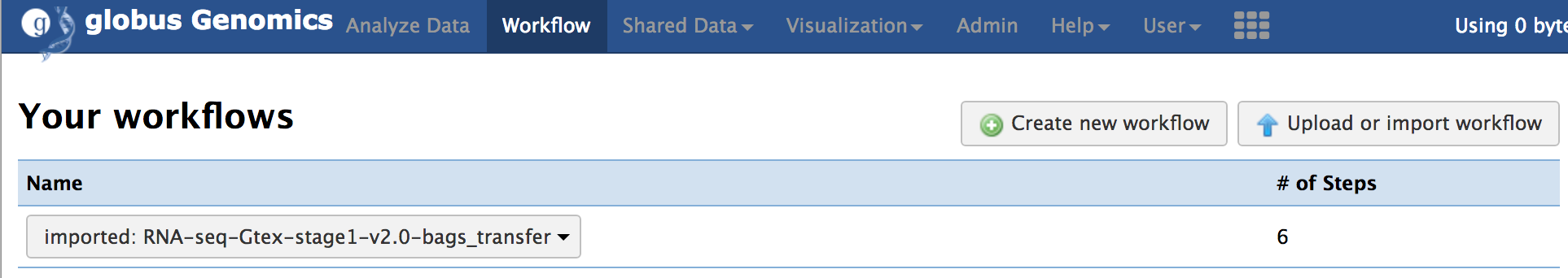
**Step-3: Import the workflow:**
From the top-menu, go to "Shared Data -> Workflows". Select the workflow named "RNA-Seq-Gtex-stage1-v2.0-bags_transfer" and click "import" to import the workflow into you account.
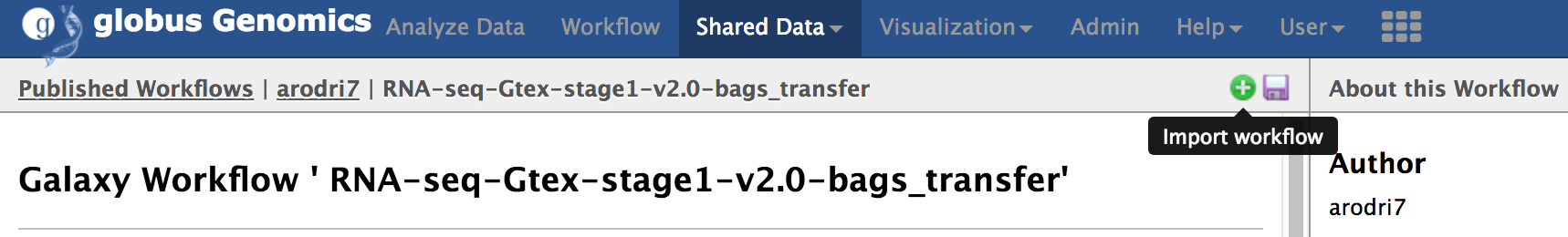
You should see the imported workflow under the "Workflow" link from the top menu.
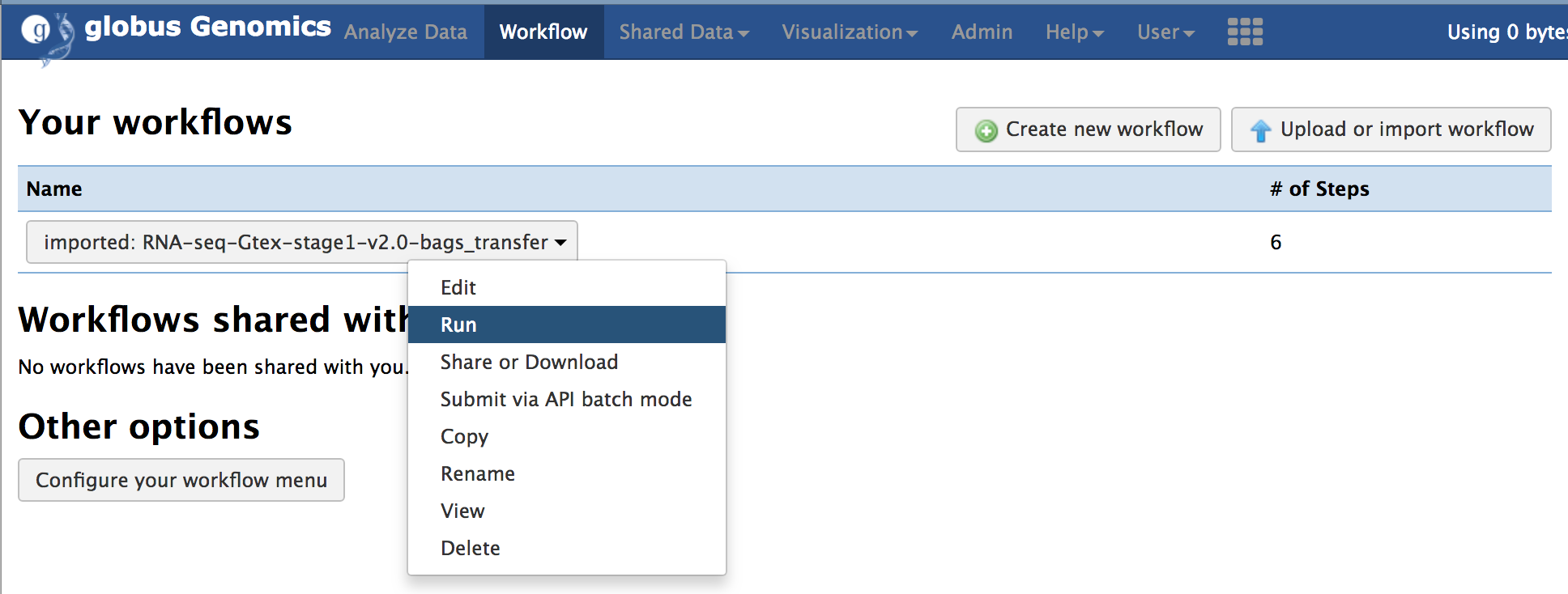
**Step-4: Run the workflow:**
Select the imported workflow from the "Workflow" menu item and click on "Run".
You will notice that the workflow only takes one input, the MINID for the [input data BDBag](#input-data): ark:/57799/b9bh8c as shown in the screenshot below:
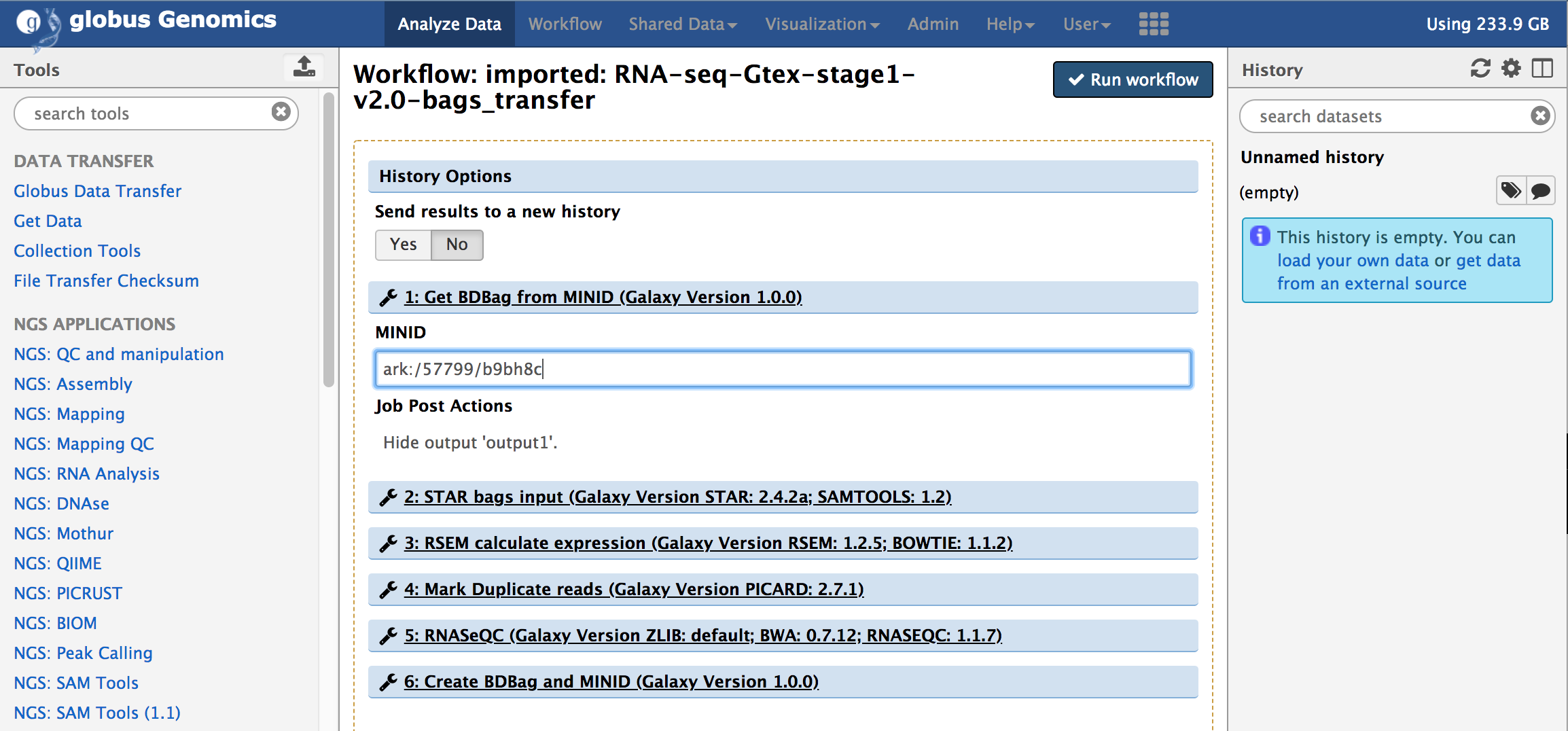
Provide the MINID in the input text box of the first step in the workflow and click on "Run Workflow".
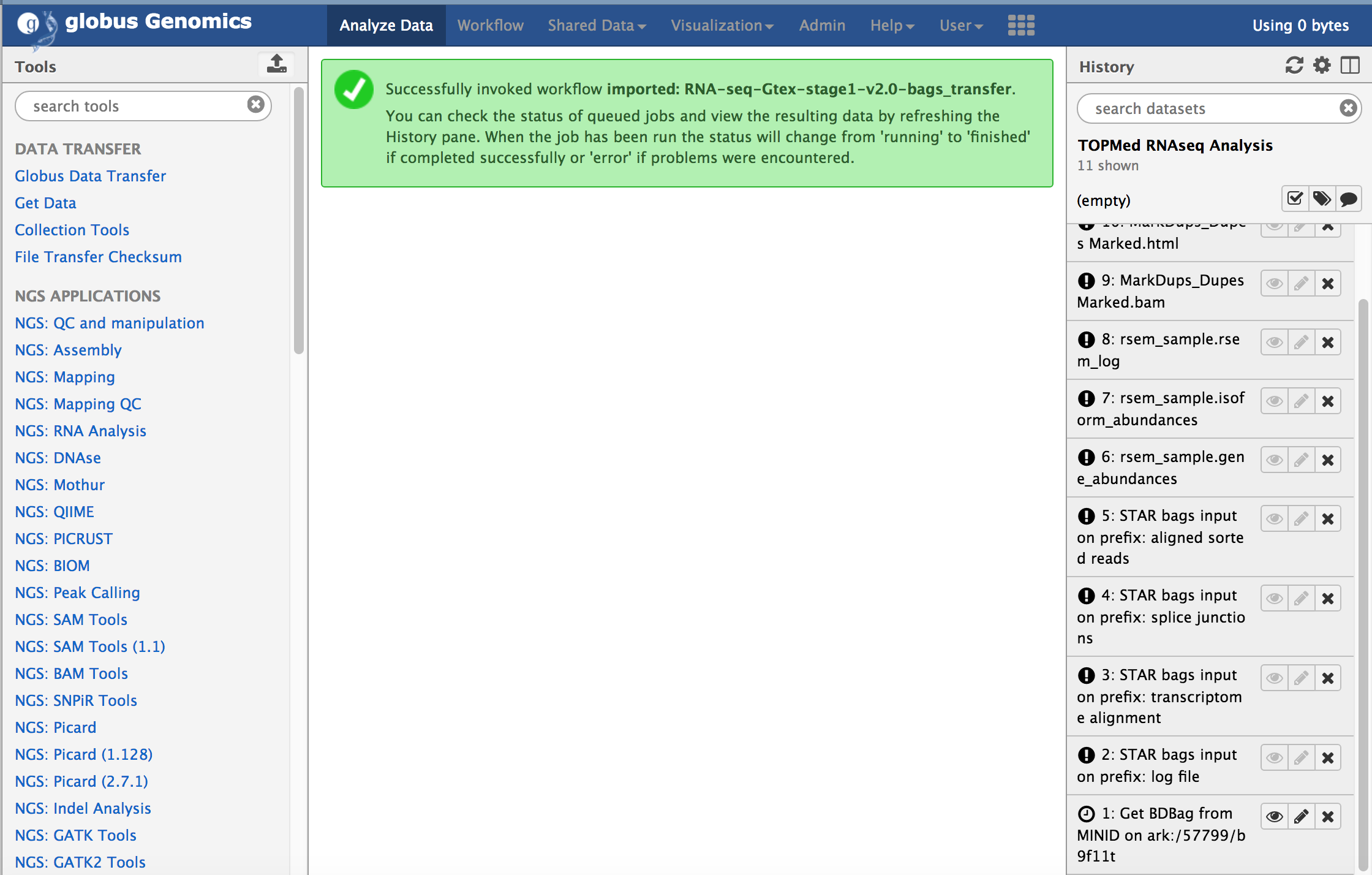
The workflow will be submitting within Galaxy and all the tasks will be added to the history. The steps within the workflow will start running one after another based on the dependencies. The first step in the workflow, "Get BDBag from MINID" will download the input sample represented by the BDBag ([ark:/57799/b9bh8c](#input-data)). In this case, it is just one sample that is download. After the download is complete, it will run all the steps one-by-one to complete the analysis and finally the "Create BDBag and MINID" step will run, that will create a new BDBag with all the outputs, provenance, and performance files in the bag.
### Validation of Results
The results of each invocation of this TOPMed RNASeq pipeline results into a BDBag that can be used to compare the validate the reproducibility of the workflow. We are providing a reference [BDBag for outputs](#outputs-provenance-and-performance) that can used to compare the content of the BDBag generated by the users. The outputs BDBag contains a "manifest-md5.txt" file that provides checksums of the outputs files generated during the analysis. The TOPMed RNA-Seq pipeline also runs RNASeQC that provides the QC results in the form of tables and graphs. These can be used to compare the reproducibility of the outputs. And example screenshot of the QC metrics is shown below:
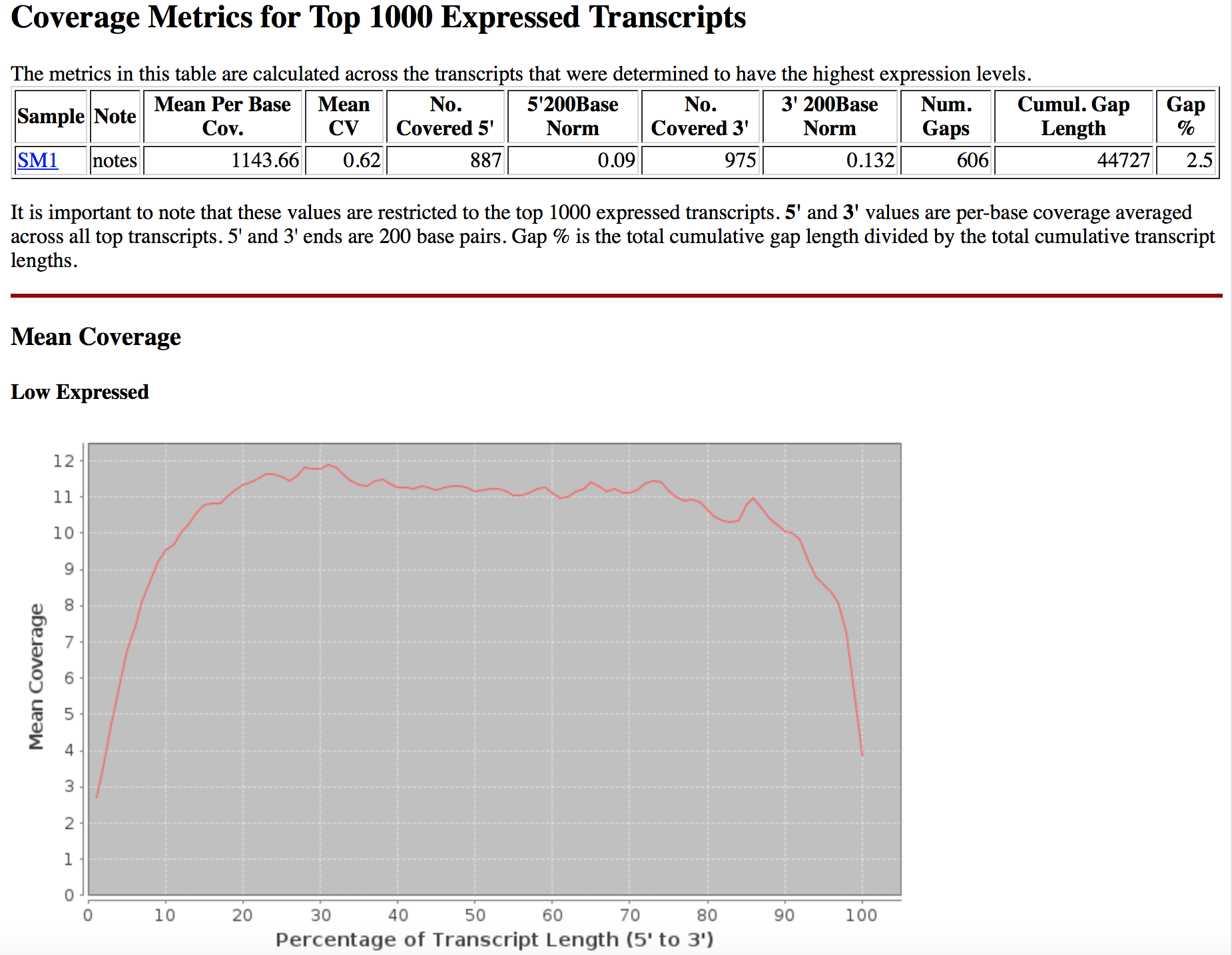
The outputs BDBag also contains a metadata file (metadata/annotations/outputs.metadata.json) that provides details of each step in the pipeline. This file includes Command-line parameters used for each tool, runtime, and start and end time of each tool. Following is an example of the information in this metadata json file:
“MarkDups_Dupes Marked.bam”: { “command_line”: “python /opt/galaxy/tools/picard-2.7.1/picard_wrapper.py -i "/scratch/galaxy/files/001/dataset_1598.dat" -n "Dupes Marked" –tmpdir "/scratch/galaxy/tmp" -o "/scratch/galaxy/files/001/dataset_1602.dat" –remdups "true" –assumesorted "true" –readregex "[a-zA-Z0-9]+:[0-9]:([0-9]+):([0-9]+):([0-9]+).*" –optdupdist "100" -j "$JAVA_JAR_PATH/picard.jar MarkDuplicates" -d "/scratch/galaxy/job_working_directory/000/763/dataset_1603_files" -t "/scratch/galaxy/files/001/dataset_1603.dat" -e "bam"”, “duration_in_sec”: 5135.991441, “job_create_time”: “2018-06-01T04:59:02.601831”, “job_end_time”: “2018-06-01T06:24:38.593272”, “job_name”: “MarkDups_Dupes Marked.bam”, “tool_id”: “rgPicardMarkDups_2.7.1” } ```
List of BDBags
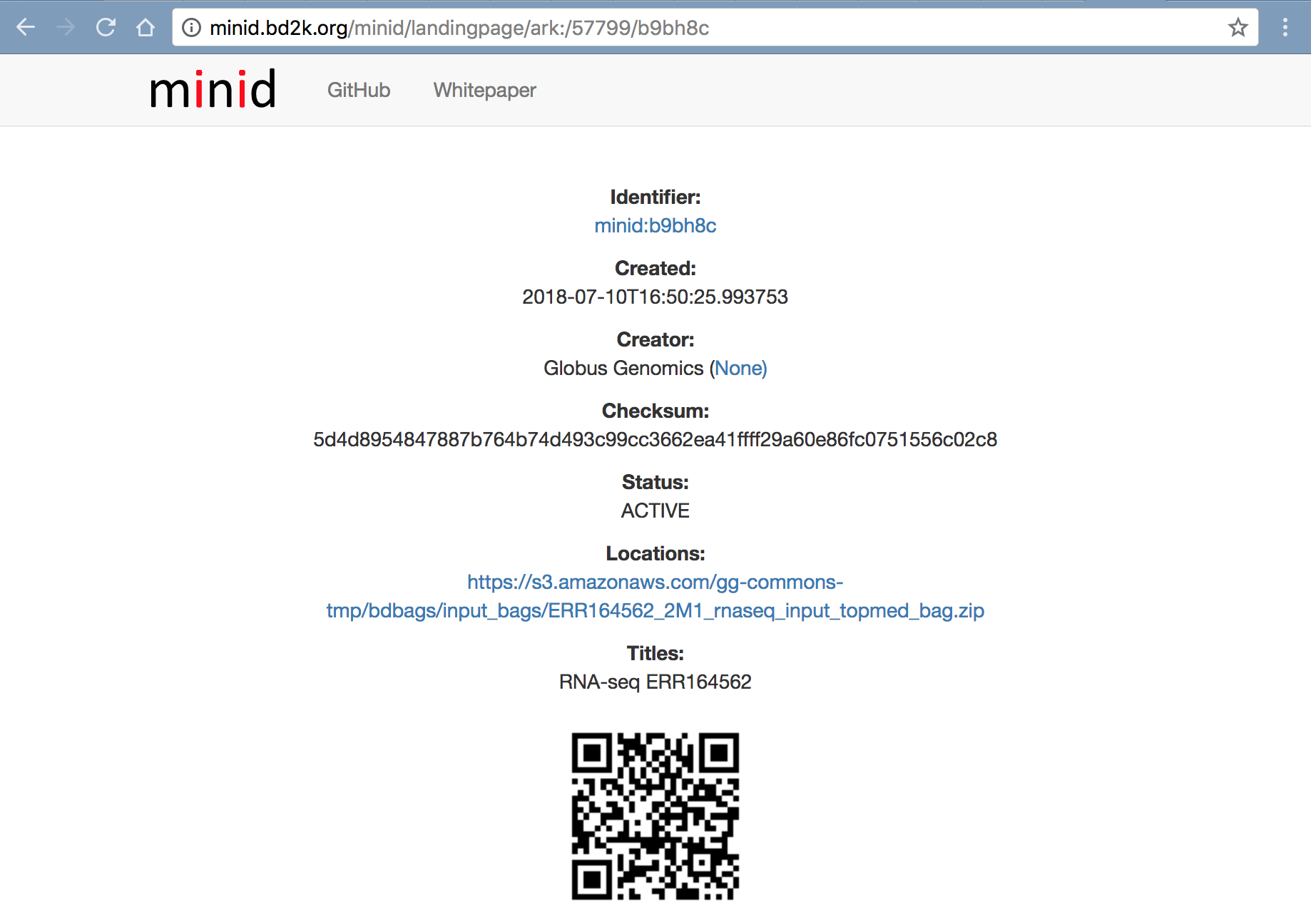
Input Data
http://minid.bd2k.org/minid/landingpage/ark:/57799/b9bh8c
Outputs, Provenance and Performance
http://minid.bd2k.org/minid/landingpage/ark:/57799/b9x12j
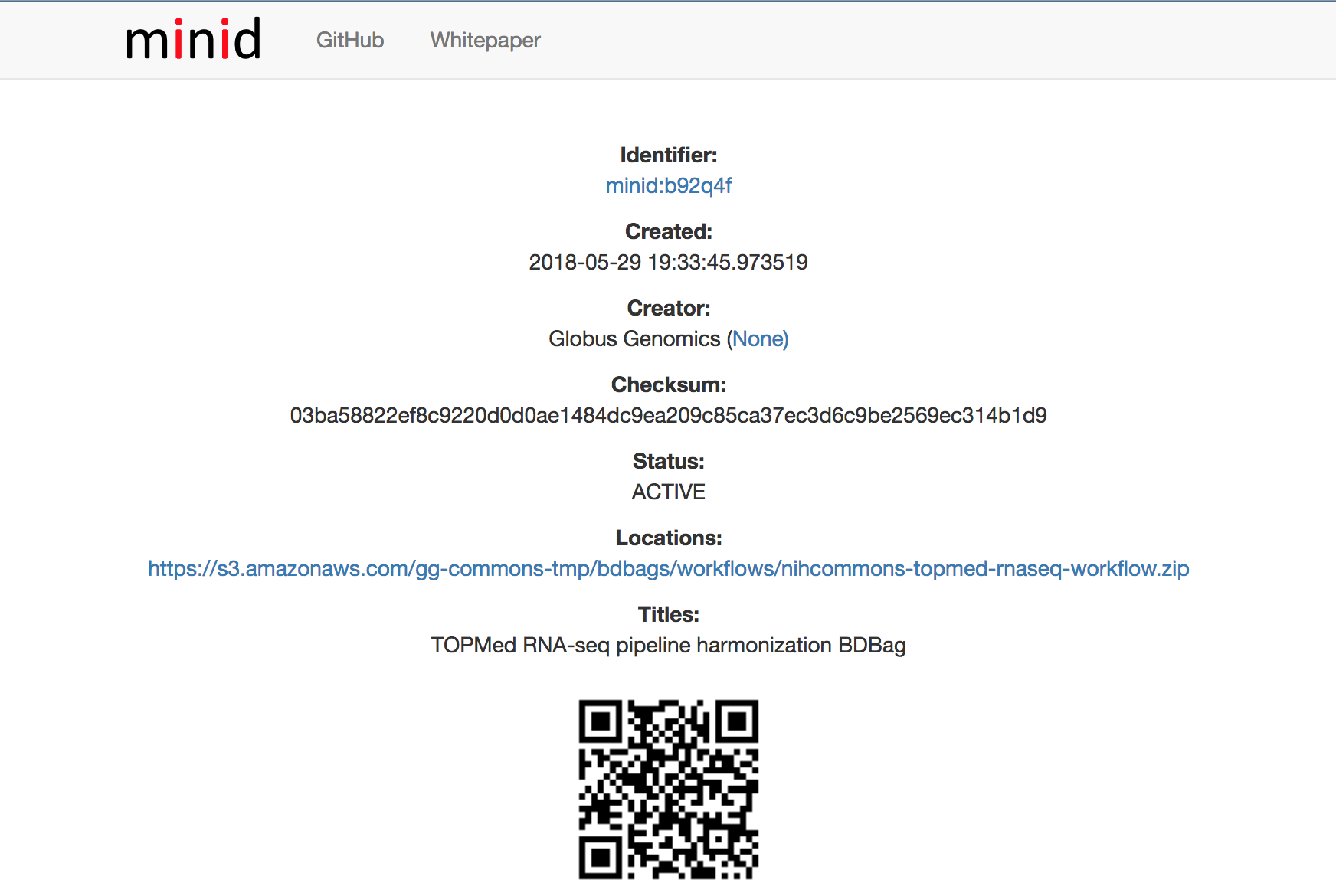
Workflow, Tools and Tool wrappers
Workflow: http://minid.bd2k.org/minid/landingpage/ark:/57799/b92q4f
Tool Wrappers: http://minid.bd2k.org/minid/landingpage/ark:/57799/b9t690
Tools reference databases
http://minid.bd2k.org/minid/landingpage/ark:/57799/b9ph5b