Rapid Annotation of GTEx Genomic Variants with the TOPMed Annotation Pipeline
Goal: Rapidly and reliably aggregate variant annotations for large volumes of whole genome sequence data, with results that are findable, accessible, interoperable, and reusable: FAIR.
Method: Leverage cloud computing and Commons tools to run the Whole Genome Sequence Annotator (WGSA) on many GTEx genomes, with identifiers, virtual cohorts, and reproducible pipelines used pervasively to ensure FAIRness.
Scalable, Cost-Controlled Analysis for Data Enrichment
Generating functional annotation of variants generated from analyzing whole genomes is an essential part of WGS data analysis. Large quantities of genome data are now available, but genome-wide association studies that depend on rare variants lack statistical power. Combining annotations from many sources provides a comprehensive “genome map” boosting statistical power of association studies. Additionally, annotations are useful to filter variants to suit the analysess of interest. For example, an analyst may be interested in variants that only enhance a particular function.
The TOPMed project uses a tool called WGSA (Whole Genome Sequence Annotator) tool for the annotation of variants generated from the project. The WGSA tool is availble as an Amazon Machine Image and is runnable from the Amazon Compute Cloud. However, it requires analysts to setup a cloud account, gain the expertise to instantiate the pipeline and assemble the required annotation databases. The default pipeline out of the box generates annotations serially using a single compute node where the pipeline is setup. This process can take weeks to months for a large set of variants and hence has the potential to drive up the compute costs of annotation.
We created a high performance, cost-effective and parallel implementation of the WGSA annotation pipeline.
The annotation pipeline uses 70 annotation databases to annotate the SNPs and Indels from 608 GTEx variant (VCF) files to create a data resource of millions of annotated variants that can then be used in genome-wide association studies. Each annotation process takes roughly 24 hours to complete on a 32-core node, for a total of roughly 500,000 core-hours. The annotation pipeline uses cost-aware provisioning to perform annotation on a compute cluster using spot instances on the Amazon cloud.
Re-Use of Data
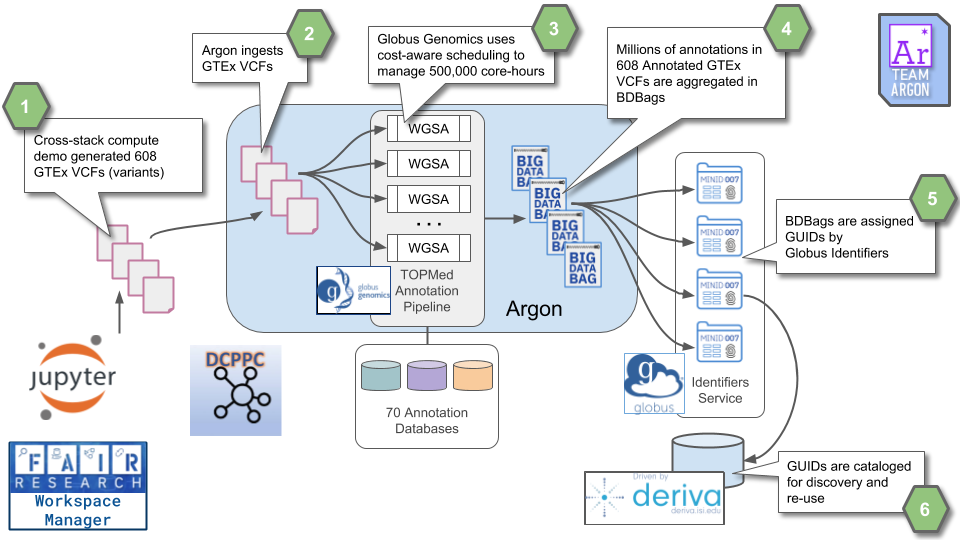
This example highlights Re-Use and interoperability of data and computational tools. The variant files come from the DCPPC Cross-Stack Compute Demo while the annotation tool is from the TOPMed project. The annotations are scaled across the input data using cost-aware cloud provisioning to create a rapid yet inexpensive pipeline. The results are packaged for sharing by using BDBags as a common data exchange format and referenced by GUIDs with agreed upon metadata conventions. Because the tools and services comprising this architecture build on interoperable components (e.g., OAuth2/OIDC used by Globus Auth) additional patterns emerge. This workflow can be driven from by a Jupyter notebook, beginning with calls to DERIVA or another portal for cohort creation, through the computation and its requisite data handling, and back to DERIVA to associate the results with the original cohort.
The Data
The annotated VCFs are being aggregated into BDBags and assigned Minids (a type of KC2 GUID, here’s an example). We’ve made an initial pull request into the DCPPC Full Stacks repository to share these identifiers with the other teams. We’ll update the new file created in that pull request (gtex-wgs-annotated-vcf-wgsa.tsv) as the annotation runs complete via additional pull requests. Also, the Minids will be ingested into our DCPPC instance of DERIVA to tie these new files back to the original sequences.
Slides
Check out this presentation for more details about the science and components.